May 21, 2024
Version 3
U2OS Nucleofection & Analysis Protocol for MSPH Validation V.3
This protocol is a draft, published without a DOI.
- Jason Waligorski1,2,
- Colin Kremitzki1,2,
- Graham Bachman1,2,
- Mallory Wright1,2,
- William J Buchser1,2
- 1Washington University School of Medicine;
- 2McDonnell Genome Institute



Protocol Citation: Jason Waligorski, Colin Kremitzki, Graham Bachman, Mallory Wright, William J Buchser 2024. U2OS Nucleofection & Analysis Protocol for MSPH Validation. protocols.io https://protocols.io/view/u2os-nucleofection-amp-analysis-protocol-for-msph-dd4828zwVersion created by Graham Bachman
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 15, 2024
Last Modified: May 21, 2024
Protocol Integer ID: 100224
Funders Acknowledgement:
Washington University in St Louis
Abstract
Validation steps.
Design
Design
Choosing synGRNA sequence
This is the method utilized in 2023 for the MSPH library in U2OS cells for validating Raft-Seq hits. This is NOT the tandem gRNA method.
- Choose one gRNA that was a hit from the primary screen, usually from a published library (Brunello)
- Design a 2nd 'backup' gRNA sequenced which is not from the same library, cross-checked with InDelphi for high frameshift %, Hi On Target, and Low Off Target
If designing from scratch, GESC recommends crispor.tefor.net and also ensure that the gRNA is in an exon that is common to as many isoforms as possible.
The majority of syn GRNAs we have tested yield >95% cutting. The fraction of out-of-frame was slightly lower.

Supplemental Figure 5. Cutting Efficiency in Synthetic gRNAs Against Genes-of-Interest.
Tandem gRNA Approach (alternate)
The approach below is published, but they use in-vitro transcribed gRNAs that are inferior to Xiaoxia's synGRNAs.
For this to work at WashU, we would need the following:
- gRNAs should be designed to have high on-target and low off-target to produce a frameshift
- gRNAs should be less than 250 bp apart so they can be sequenced in the same amplicon
- gRNAs should be in an exonic region, and not span introns or intron-exon boundaries
- Cut site distance should not be divisible by 3 . . even so, it is possible to get an in-frame deletion because of INDEL occurring when the two sides recombine
- The Paper shows gRNAs have to be ~50 bp or more separated to be effective
- Of note, although the synergistic benefit is high with further separated gRNAs, that absolute cutting efficiency is sometimes low (EPHX2 is worst case, but even combined ADK is worse than we usually get with a single gRNA)

Synergistic effect of tandem guide RNA (gRNA) combinations. (A) Bar chart displaying the percentage of remaining wild-type alleles in CRISPR/Cas9 gene editing (GE) experiments in HepG2 cells using either each gRNA alone—the most efficient guide (driver [D]), the second guide (helper [H]), or the synergistic tandem combination (T)—for 12 targets (14 combinations). An expected additive gRNA combination has been calculated (see Methods) and is also displayed. Distance between the two Cas9 sites is indicated inside brackets. No synergistic effect is observed when the two sites are too close. Horizontal bars: 50% and 90%. (B) Scatter plot showing the synergistic benefit (calculated as the difference between the %GE obtained with the tandem synergistic approach and the %GE that would be expected if the effect of the two gRNAs would only be additive) in relation to the distance in between the two Cas9 sites. Green dots, positive synergistic benefit; red dots, negative synergistic benefit. ADK-S, 1 bp in between Cas9 sites; ADK-L, 58 bp; FABP-S, 26 bp; FABP-L, 52 bp. Horizontal bar, 35 bp.
Ordering synthetic sgRNAs
Ordering synthetic sgRNAs
Note:
In addition to ordering synthetic gRNAs of interest, synthetic gRNAs should also be ordered as controls. Using endogenous mutants, or any cell line that does not undergo electroporation (even if it is electroporated without a synthetic gRNA) is untenable for the purposes of investigating subtle image-based phenotypes in cell populations containing a gRNA of interest. This is because the difference between exogenous and endogenous cell lines of the same genotype is often more pronounced in images than the subtle phenotypic differences that manifest from different gRNAs.
Click “PRODUCTS & SERVICES” -> “CRISPR-Cas9”

Scroll down to “ORDER IN TUBES,” click.
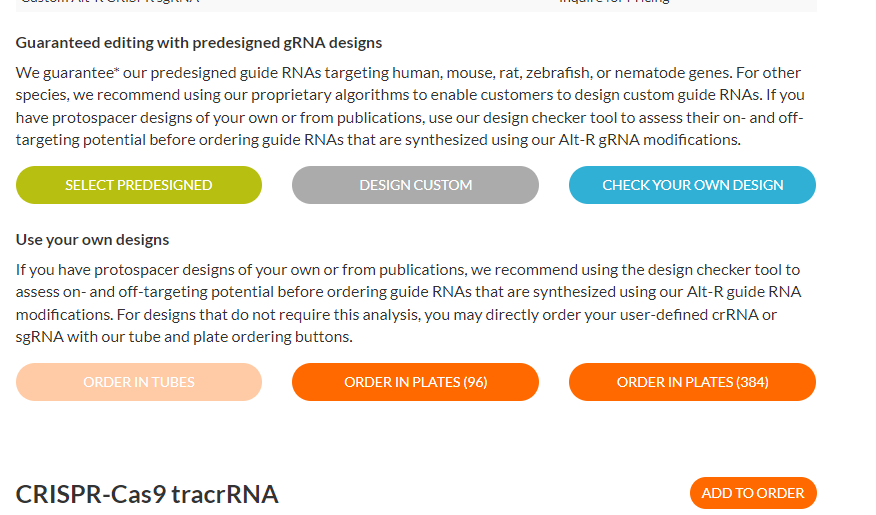
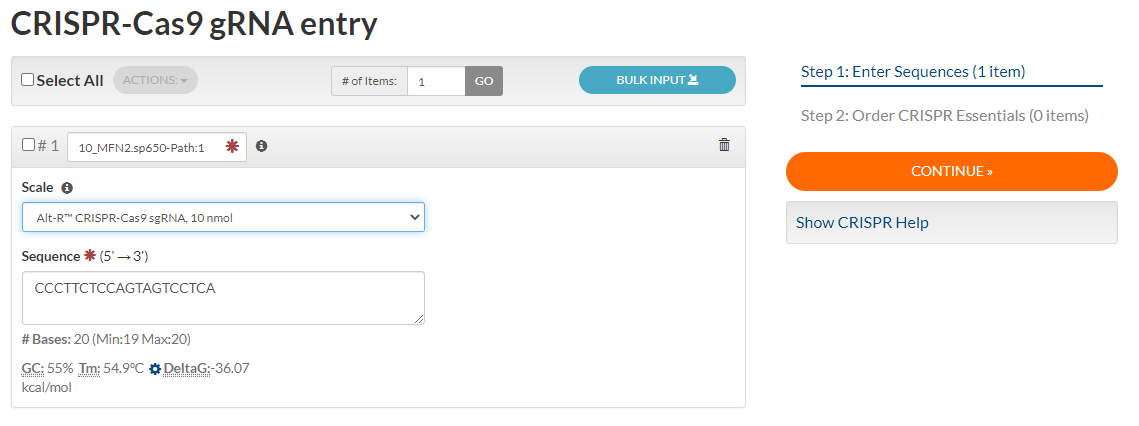
Input the full name of your gRNA into the input field next to #1. Select “Alt-R CRIPSR-Cas9 sgRNA, 10 nmol” from the “Scale” drop-down menu. Type your full 20 base sequence into the “Sequence” text box. Select the desired quantity (1 = a single tube). Click “CONTINUE”. Check if we are out of Nuclease-Free Duplex Buffer, add to order if necessary (see step 8). Select “ADD TO ORDER.”
Click “CONTINUE SHOPPING” to add any additional synthetic gRNAs to your order. When you are done, click “CHECK OUT.”
Under “Billing Address,” change the drop-down menu to “Accounts Payable.”
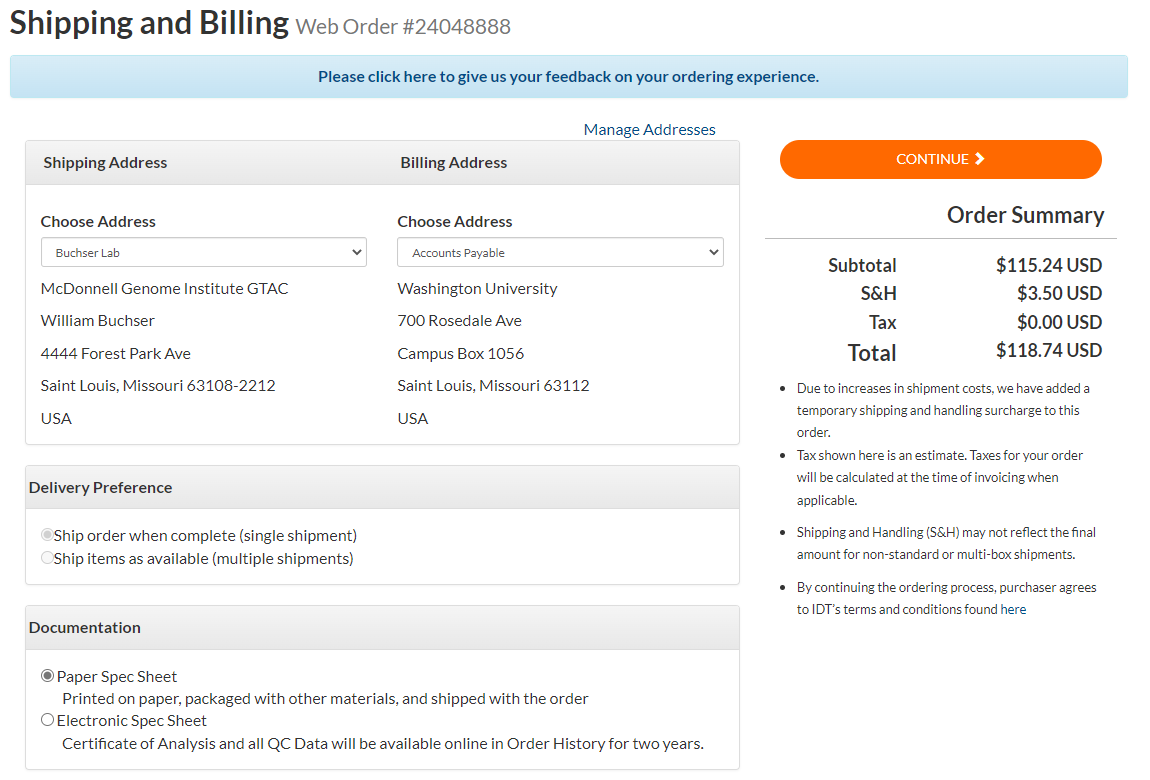
Change the “Choose Payment” drop-down menu to “Buchser,” other fields will automatically populate. Click “SUBMIT ORDER.”
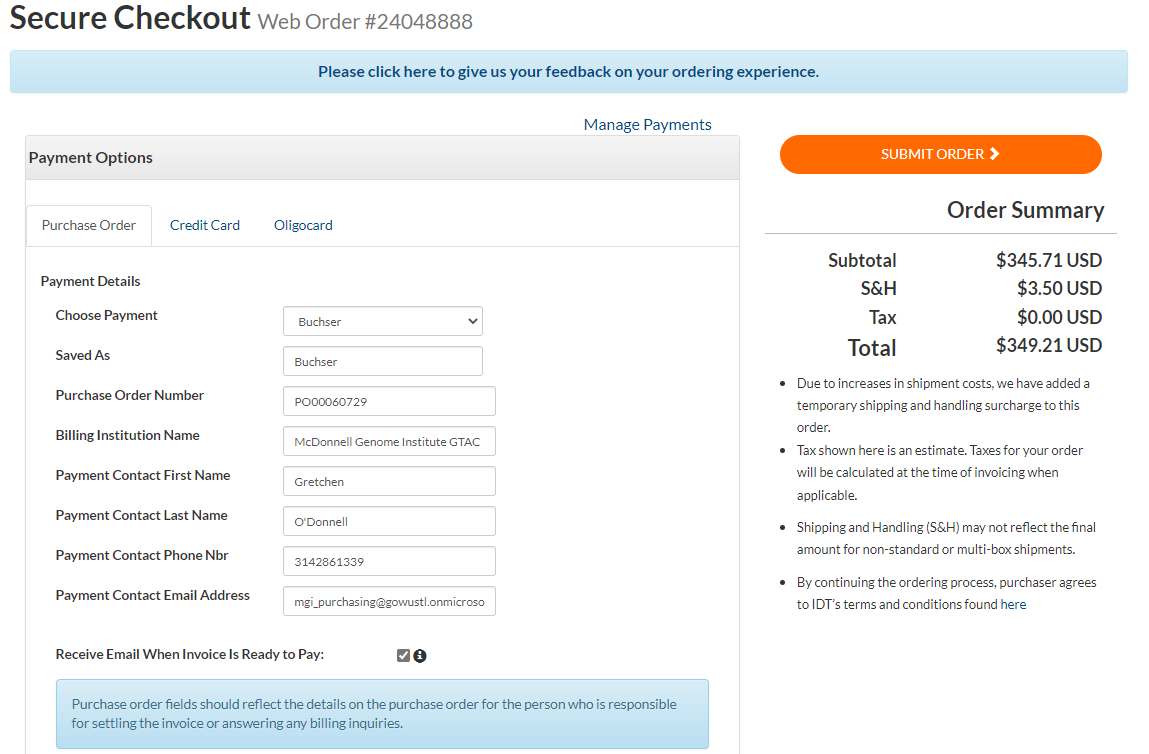
When your synthetic gRNAs arrive you need to dilute them in a Nuclease-Free Duplex Buffer. There should be a bottle in the “Mariel” 4 degree. If necessary, you can order a new bottle from the “Order CRISPR Essentials” page after you enter the synthetic gRNA sequence.
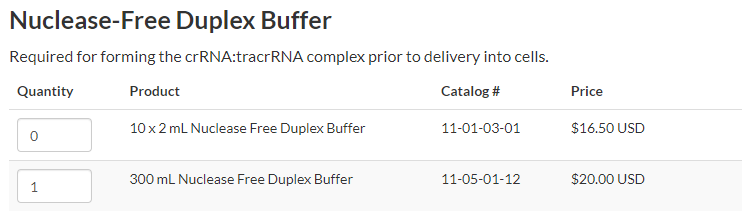
Dilute each synthetic gRNA in n microliters where n is 10 times the stock concentration. For example, if your stock concentration is 10 nmol, dilute the synthetic gRNA in 100 μl.
RNP Nucleofection
RNP Nucleofection
How we nucleofect LUHMES : dx.doi.org/10.17504/protocols.io.36wgq32d3lk5/v3
Protocol

NAME
CRISPR/Cas9-Mediated Knockdown in LUHMES Cells: Nucleofection and Validation ProtocolCREATED BY
Mallory Wright
8 nucleofections are run in parallel on a 16 well cuvette (Lonza strip) and plated onto 12-well plate(s).
A single experiment will produce 4 12-well plates, each with 8 experimental synthetic gRNAs, 2 negative controls and 2 positive controls 3 Lonza strips.

An 8-tip INTEGRA is needed for transferring aliquots from the 16-well Lonza strip to a 12-well plate. See graphic below:

Complex 2µl of synthetic gRNA with 1µl of Cas9 protein in a 0.2ml snap tube for each condition and label (snap tubes should be arrayed in column(s) of 8).
Complexed RNPs can be stored at -20C for several weeks without degradation or made day-of. Either way, place on ice prior to nucleofection.
Mix Lonza primary solution with supplement (both stored at 4C) at an 82:18 ratio and place on ice. This is the P3 solution.
The amount of P3 solution required is determined by
(17 * n) + e
where n is the number of wells to be seeded, and e is an additional amount of P3 solution in µl, added to ensure no issues in aspirating from the INTEGRA boat later.
e was 40µl throughout MSPH validations.
The total volume of a single cuvette well is 20µl, 3µl of which is taken up by the RNP, thus the constant of 17µl.
Place 2ml of McCoy’s Tet-Free FBS Media in each well of the 12-well plates and place in 37C/5% CO2
incubator.
Thaw parental cell line or take from existing flask: U2OS iCas9 (3e5). Count cells and take aliquot(s).
The size of the aliquot is determined by the number of wells (n), the target density (d), the amount of additional P3 solution (e), a fudge factor (f), and the average cell count per µl (µc).
A = (f * d * ((d * n) / (17 * n) * ((17 * n) + e) / (d * n)) / µc) * n
n should be no more than 8 to ensure parallelization between conditions.
In our experiments: d = 75,000; e = 40 (step 14); f = 1.1 and µc was averaged from two counts.
This excel sheet may be useful for calculations:
| A | B | C | |
| N Well | 6 | ||
| Target Density | 75,000 | ||
| Additional P3 Solution | 40 | ||
| FF | 1.10 | ||
| Count A | 574,000 | ||
| Count B | 524,000 | ||
| Cells per uL | AVERAGE(B5:B6)/1000 | ||
| Resuspned in y P3 | ((17*B1)+B3) | ||
| Total Cells Needed | (B2*B1)/(17*B1)*((17*B1)+B3) | ||
| Density Factor | B9/(B2*B1) | How much to multiply target density by to ensure retention of target density when resuspending in larger volume of P3. | |
| Working Density | (B2*B10)*B4 | Density to use in calculation with cell count | |
| Final volume | (B11/B7)*B1 | How much cell solution to bring into 1.7ml snap tube |
Centrifuge aliquot at 1200 rpm for 3 minutes and aspirate media, being careful not to disturb pellet. Resuspend aliquot in PBS (washing media off the cells). Centrifuge at 1200RPM for 3 minutes and aspirate PBS, being careful not to disturb the pellet.
Set up the Lonza 4D Nucleofector: 1. Select "X,"; select the 16-well cuvette icon; select the wells to be electroporated; select P3 for cell solution; input DS-150 as the pulse code.
Remove 12-well plate with warmed media from incubator.
P3 solution is toxic to cells, so minimize time that cells are in this solution.
Resuspend aliquot in P3 solution and transfer to INTEGRA boat.
Using an 8-tip INTEGRA, gently and briefly (1-2 aspirations/dispensations) mix the cell solution from the INTEGRA boat with the RNP complexes in the 8-strip of 0.2ml snap tubes.
Aspirate and transfer to one of the columns of the Lonza strip.
Transfer strip to Lonza 4D Nucleofector machine and press start. The machine will automatically eject the strip when complete.
After nucleofection, return Lonza strip to hood and neutralize P3 solution with a few µl's of warmed media.
With the 8-tip INTEGRA, loaded with a tip on every other terminal, aspirate 4 of the cuvettes and transfer to 4 wells in one of the rows of the 12 well plate. Repeat to plate the second row of the 12-well plate.
Adjust the INTEGRA spacing between aspiration and dispensation. On a 1125µl INTEGRA, use 6mm spacing for aspiration and 14mm spacing for dispensation.
Cells are prone to clump together after plating. To help ensure the cells take advantage of the full surface area of each well, triturate the cells against the wall of each well repeatedly and then move the whole plate around in a figure eight motion.
Once the cells have been plated, avoid transporting the plate to and from the incubator more than necessary. Media sloshing about tends to move the cells back to clumps. Therefore, when transporting the plate, do so gently, keeping the plate as level as possible.
Repeat steps above as many times as needed.
Cells should be monitored daily over the next week. Consider a media change 1 day after nucleofection (especially if there is significant cell death).
A media change should occur 4 days after nucleofection.
After one week, cells should be ready to be re-plated into 96-well optical plates for imaging.
Plate Randomization
Plate Randomization
Generate plating files:
Open FIVE Tools and navigate to "Layouts" window.

Fill in the details for your plate.
Double check all settings, but you should expect to update: Exp Name, Export Folder, Cells per Well, and the table to the right of the window.
An example of a properly completed Layout is shown below.

Once details have been filled in, click "Fill Plates" and then "Export This Arrangement."
Setup BioMek:
On the BioMek PC, navigate to "BioMek working protocol."

Should look like this:
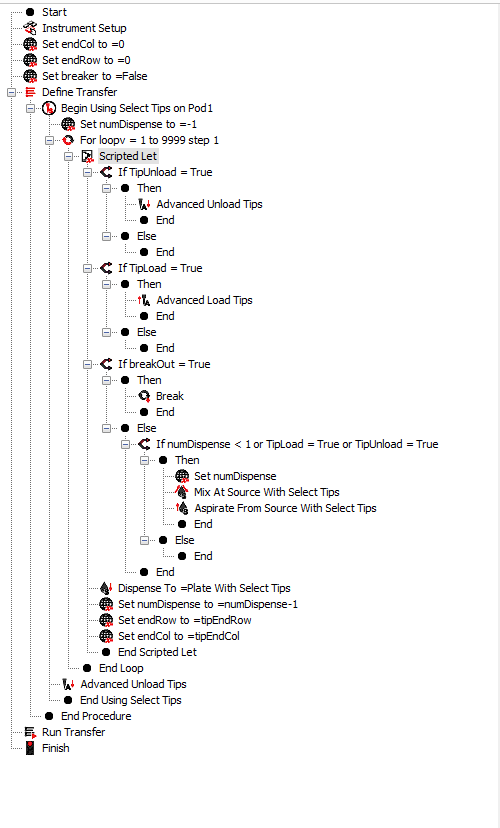
Ensure the BioMek deck is arrayed as shown below:

- empty_2 and empty_1 should contain empty tip boxes. Ensure the boxes are completely empty before starting.
- tip_box should contain a tip box with at least 24 tips in the bottom rows, starting from the bottom right. tip_box can contain more tips, but the BioMek arm begins from the bottom right and does not snake, so at minimum ensure tips are present in the bottom rows and right-justified.

- "0" and "1" should contain 96-well optical plates. "2" and "3" can be left empty.
- "Source" will contain a new 12-well plate with cell solution.
Seeding the source plate and running randomization:
For each 12-well plate, aspirate each well and add 200µl of trypsin and return plate to 37C/5% CO2 incubator. After 5 minutes, remove plate(s) from incubator and neutralize trypsin with 1ml of McCoy’s Tet-Free FBS Media.
Transfer the contents of each well to a labeled 1.7ml snap tube. Spin down, aspirate supernatant and resuspend cells in 500µl of media.
Prepare to take cell counts. This step is especially time consuming. You may consider grabbing a lab mate to speed up the process and minimize the number of times you break the sterile plan of the hood to record counts.
In Excel, open the FIV###_PlatingSetup.txt file generated from "Exprt this Arrangement" in the FIVE Tools Layout window. Add two columns to the table, "Count A", and "Count B." Update the formula in the Cells/uL column to be the average of these the values in these two columns for each row.
Begin taking cell counts, updating the spreadsheet as you go. The columns "uL Media" and "uL Cells" tells how much media and how much cell solution from your 1.7ml snap tube to add to each well of the new 12-well "source" plate.
Add correct amount of media to each well of the source plate and add the appropriate number of cells. Any remaining cells left in the 1.7ml snap cap tubes will be saved for NGS sequencing. Write the Cells/µl count on the side of each tube. See PCR and NGS Submission/Analysis section below how to properly process the remaining cells.
Load source plate onto BioMek, ensure all lids are removed and tips are properly stocked.
Replace the text in "Scripted Let" with the text from the FIV###_script.vb file generated from the "Export This Arrangement" button in FIVE Tools Layouts window.
Select "Run."

Select "OK."

When finished, transfer 96-well optical plates to incubator. Cells should be plated for ~16 hours before staining (usually the following morning).
Staining and Imaging
Staining and Imaging
96-well plates are to be scanned on the HT Ai confocal microscope, but can be adjusted for any confocal microscope that can detect Blue, Red, and DeepRed channels.
Make a staining media by combining the following dyes as such in a compatible cell media (McCoy's Tet-Free FBS Media for U2OS cells):
Hoechst 1:2500ul media
TMRM: 1:1000ul media
Mitotracker DeepRed: 1:1000ul media
Aspirate existing plate well media and add 100ul of stain media. Allow plate to stain in a 37C/5% CO2 incubator for 30 minutes.
Aspirate stain media and wash each well with 100ul of media and return to incubator until ready to image.
Set up HT Ai to collect the Blue, Red, DeepRed, and Brightfield channels and focus on the plate bottom.
Dial in Exposure, Laser power percentage, and Focus of each channel and collect as many images as needed. Make sure to label images properly for retrieval after scanning is complete.
PCR and NGS Submission/Analysis
PCR and NGS Submission/Analysis
Now that you've Plated, Stained, and Imaged the cells, it's time to process your cells for Next Generation Sequencing. At the time of plating, you should have saved the remaining cells for each synthetic gRNA. These will be used to test the cutting efficiency of Cas9. These cells should be in properly labeled 1.7ml snap cap tubes with cell counts. Centrifuge these tubes in a centrifuge with the snap cap tab in the top position. This will allow you to visually see your cell pellet, or know where the cells should be if your count is low. Centrifuge for 3 minutes at 1200 rpm to pellet the cell and to remove supernatant, cautious not to aspirate the pellet. Cells should then be resuspended in Extraction Buffer at a ratio of 100ul/500K cells and processed through two pre-warmed heat blocks. 65C for 15 minutes, 95C for 5 minutes, then 4C storage. The following protocol is listed for making Extraction Buffer and to properly extract the DNA (https://docs.google.com/document/d/1jzNhZdHz9bz731jB_lTLPowFR0hkQPq9bEsJVN6YzfQ/edit#heading=h.lzms4duz8cm3)
Utilize the NanoDrop one to determine your DNA quantity before taking samples into PCR1 and normalize DNA concentrations from 100-500ng/µl if possible. If there is high protein concentration in the samples or if the supernatant is viscous, consider adding more Extraction Buffer and reprocessing to degrade more protein.
Make sure you have primers ordered that flank the gRNA cut sites. We order three different 20-mer primer sets, not to exceed 300bp amplicons in length, and run them against generic bulk DNA to test which primers work best. Ideally primers should flank the gRNA ~150bp on each side, but you might not be able to get the gRNA in the middle of the amplicon. As long as the primers don't overlap the cut site, you should be good. See Primer Design and Ordering Protocol on how best to select primer pairs. Primers should have the following DeepSeq tags in order to multiplex samples on the Illumina Sequencers. We will link well specific Index Primers in the PCR2 section. You can follow the PCR1 and Gel Confirmation protocols below to test the primers and to run your actual samples. Select the primer set that produces the brightest single band in gel verification.
The two DeepSeq tags are (5’ -> 3’):
Forward: CACTCTTTCCCTACACGACGCTCTTCCGATCT
Reverse: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
So instead of ordering the forward primer CGCTCTGCTAGCTATCCCTG and the reverse primer GCTCGAGGGATCCGTTAACTC, we would order CACTCTTTCCCTACACGACGCTCTTCCGATCTCGCTCTGCTAGCTATCCCTG and GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCTCGAGGGATCCGTTAACTC
DeepSeq primers are only used in the PCR1 section. Forward and reverse Index primers are assigned to us by the Sequencing Core and are specific to our lab. Index primers are attached in PCR2.
PCR1 MasterMix recipe and Cycler conditions:
Prepare the PCR reaction mix in a sterile microcentrifuge tube by combining the following components:
Template DNA: The DNA you wish to amplify, need ~100ng of DNA per 25µL reaction
Forward and Reverse Primers: Short DNA sequences that bind to the start and end of the target DNA region you want to copy.
MyTaq HS Red Mix (Meridian Life Science Catalog #BIO-25047): A ready-to-use PCR master mix containing DNA polymerase, dNTPs, buffer, and a red dye.
DMSO (Dimethyl Sulfoxide): Added to enhance PCR specificity and amplification of GC-rich templates
Ambion Nuclease-free Water (Thermo Fisher AM9937): added to bring reaction volume to 25µl
| w/ 100µM Primers | +10% Extra | |||
| # of reactions | 1 | 10 | 844.8 | |
| Fwd (100µM) | 0.1 | 76.8 | 84.48 | |
| Rev (100µM) | 0.1 | 76.8 | 84.48 | |
| MyTaq | 12.5 | 9600 | 10560 | |
| DMSO | 1.25 | 960 | 1056 | |
| Nuc Free Water | 9.05 | 6950.4 | 7645.44 | |
| Template | 2* | |||
| Total Rxn Vol. | 25 |
Always make 10% more of Mastermix to account for loss in reservoirs and pipette tips. Since you will most likely be testing multiple sgRNA Cutting samples, you will need to make gRNA specific Mastermixes. Ideally you will have tested all three primer sets and chosen the best one, because making mastermixes for three different primer sets for each sgRNA will get out of control very quickly. Take extra care to not mix up mastermixes or templates as it can be very confusing. Normally we do three reactions for each primer set, with one water as template negative control well. Add mastermixes to the wells first, then follow with your templates. Seal the plate with an adhesive plate seal and spin down to collect samples. Run on the following cycler program made specifically for BULK PCR1 samples.
*If more or less Template needs to be used, subtract it from the water in each reaction.
PCR1 Cycle conditions for Bulk Cellular DNA template - 35 cycles
95C - 2:00 min
95C - 15 sec
57C - 15 sec
72C - 30 sec
Repeat above 3 temps 34 more times
72C - 2:00 min
4C - forever
Agarose Gel Confirmation:
Once PCR1 has been completed, follow the Agarose Gel Confirmation Protocol below to see if you have amplicons for each validation. Set up a gel tray with combs in a gel cassette holder so that it is completely sealed and there won’t be any leakage
- Set up cassette with combs in gel rig container so that it is completely sealed and there won’t be any leakage
- Weigh out 1g of GP2 agarose (MidSci - #BE-A125) for a 2 comb gel or 2g for a 6 comb gel and add to an Erlenmeyer flask. Pinch the weigh boat for easy addition to the flask
- Add 0.5% TBE buffer to Erlenmeyer flask. The amount varies based on the size of the gel - 70ml for a small 2 comb gel, or 140ml for a large 6 comb gel
- Microwave until all agarose dissolves and the mixture is pretty much clear - 1min 15sec for 70mL, 1min 25sec for 140mL and higher (though this isn’t exact)
- Grab the flask from the microwave USING GLOVES/HOT HANDS and run the outside of the flask under cool running water for a bit
- Add SYBR-Safe dye to the flask. The volume added is 1/10000 the volume in the flask, so 70mL liquid gel => 7µL SYBR-Safe. Swirl until the dye is mixed
- Pour the gel into your gel rig and wait until cooled (~30 min). Check for leaks
- Once set, place the gel and cassette into the gel box containing 0.5% TBE buffer and remove the combs carefully
- If you look from the side, the buffer should go at least a centimeter above the gel
- Using the rectangular holes left by the combs, pipette 5µl of 100bp ladder (GoldBio - #D001) into wells flanking your samples and then 5µl of your samples into the rest
- Attach the electrodes (RED at the bottom, BLACK at the top; you can remember this by “Black in Back” or “Run towards Red”)
- Set the voltage to 170V, the mAmps to 400, and the time to run 24 minutes and hit start
- Make sure there are bubbles being formed by the front and back wires in the buffer
- After 24 minutes ake the gel out and image it in the GeneSys InGenius 3 UV chamber, usually around 240 exposure
- Save image into the Buchser Lab Gels folder by pressing the “Export For Publication” tab at the bottom and exporting it as a .tif file
- Name the file in this format: YYYYMMDD_FIV###_AdditonalDetails_Initials
Multiplex Indexing Scheme, PCR2, and Pooling:
This is the step where we add indexes samples and pool them for NGS submission. Technically the PCR2 and pooling/cleanup steps are separate, but we have usually done them in quick succession for convenience.
Our input for PCR2 is PCR1 product, importantly containing the universal DeepSeq tags. The DeepSeq tags are what the forward and reverse primers containing the all-important indices (aka barcodes) latch onto in order to do the overhang PCR.
Once the indexes are added via PCR2, we can simply pool the PCR2 products together. In order to go from pooled samples to an actual NGS submission, we have to clean up the product using magnetic beads and dilute it to a standard molar concentration.
The Buchser Lab Index Scheme
(This system was largely copied from GESC, because it works pretty well)
Our system of Barcoding samples uses pairs of indexes, a forward (6bp) and a reverse (10bp). Adding different barcodes according to sample allows us to combine multiple samples together (multiplexing) which makes things a lot more efficient. The indexes are surrounded by sequences called adaptors to make the full primer. The adaptors are for binding to the DeepSeq tags on one end and binding to the oligos in the Illumina sequencer on the other end. Here is an example of the forward index primer used in PCR2 (index in red):

You can find the forward indexes here:
R:\FIVE\Genotyping\Dual Index Primers FIVE.xlsx
And the reverse index primers here:
\\genstorage.wustl.edu\smlab\Protocols\NGS in Index_Plate_Ordering_Template_PZ.xlsx
for Z=1,2,3,4
Note: The reverse indexes are the variable region within these primers, but they are reversed-complemented from the actual index sequence that the SIC uses.
We share the reverse indices with GESC, but the forward indices are assigned solely to the Buchser Lab, so be careful in giving those sequences out.
There are 16 different forward indexes (NGS.1 - NGS.16) and 384 different reverse indexes (Plate1_A1 - Plate4_H12). The forward index primers are just in different tubes, but the reverse index primers are organized into 4 different 96 well plates (hence the naming scheme), so that instead of pipetting out from 384 different tubes, we can use the Biomek i5 that will take a sample from a whole 96 well plate 4 times. The forward index file also shows how we number plates for NGS, e.g. NGS.1 with the Reverse Index Plate 2 is Plate101 and NGS.5 with the Reverse Index Plate 3 is Plate118. These plate #s are what show up when we get our sequencing results back.
What matters is the unique combination of indexes, so we can pool up to 16 x 384 = 6144 samples = 64 96-well plates of samples without any overlap.
PCR2 Mastermix recipe and Cylcer conditions:

Make the PCR2 Mastermix consisting of Forward index primers, MyTaq polymerase, and Nuclease Free Water. Reverse Index primers can be added by hand or by Biomek to each well, as well as the template from your PCR1 plate. Run the plate on the following cycler program:
PCR2 Cycle conditions for Bulk Cellular DNA template
95C - 2:00 min
95C - 30 sec
56C - 30 sec
72C - 40 sec
Repeat above 3 temps 4 more times
72C - 2:00 min
4C - forever
Ampure Bead Sample Cleanup:
Once PCR2 has completed, your samples can be pooled by hand into one bulk sample. We then clean up the sample with the following protocol before submission to the Sequencing Core
- Label one 0.2mL tube with 0.6
- Transfer 100µL of the NGS pool into the 0.2 mL tube.
- Obtain Ampure XP beads from the fridge and vortex until all beads are in solution
- Dispense 60ul of beads into the 0.6 tube
- Pipette thoroughly to get a homogenous mixture of beads/sample
- Incubate at room temperature for 5 minutes.
- After incubation, place tubes on 96-well plate magnet for 3 minutes
- Remove 155ul supernatant and discard in labeled waste container. Leave tubes on the magnet.
- While on magnets, dispense 200µL of 70% ethanol in each tube. Incubate for 30 seconds. Aspirate 200µL of ethanol from each tube.
- Repeat for a total of 2 washes
- Use a 10ul pipette to remove an residual ethanol from well
- Remove tube from the magnet and dispense 40µL of water in tube. Triturate to get a homogenous mixture. Incubate for 3 minutes.
- Place the tube back on the magnet. Incubate for 1 minute or until the solution becomes clear.
- While on the magnet, carefully transfer the supernatant to a new 0.2mL labeled tube while avoiding any of the beads on the side.
- Calculate the concentration (ng/µL) using GESC’s nanodrop instrument.
- Select dsDNA.
- Wait for instrument to Self Test.
- Add 1.2ul Elution Buffer.
- Wipe away sample and add 1.2ul Sample.
- Record reading which usually should be between 10ng/ul and 60ng/ul
- Perform a dilution using the nM Conversion Calculator for NGS spreadsheet on the desktop of the computer next to the nanodrop instrument.
- Enter the concentration(ng/µl), BasePair Length= Amplicon size. Then look at the returned information in the dilution column. This is the amount of your cleaned up sample you should add to a labeled 1.7mL tube. The total volume of the tube should be 40µL. For example, the spreadsheet returns a dilution value of 2.58. In the 1.7 mL tube, you will add 2.5µL of the cleaned up sample to 37.5µL of Elution Buffer.
- Label the 1.7mL tube with colored dot with Buchser Lab, Sample Name, 2x150 run, Date and Initials
- Send the Genotyping Submission Form to CGSSB-DSIL@LISTSERV.WUSTL.EDU
- Take the tube to Jess and MariaLynn on the 4th floor of the Couch building. If it’s Tuesday afternoon, drop tubes off in Jess’s Ice Bucket on her bench. Any other drop offs can go in the small drop off freezer in the equipment hallway.
Once your NGS reads that have been returned from the Sequencing Core, you can follow the protocol below to download the NGS reads and run cutting analysis.
Protocol

NAME
Library Aligner and NGS Cas9 Cutting AnalysisCREATED BY
Colin Kremitzki