Mar 21, 2022
Version 2
Plant assemble - Plant de novo genome assembly: annotation V.2
- Scott Ferguson1,
- Ashley Jones1,
- Justin Borevitz1
- 1Australian National University



Protocol Citation: Scott Ferguson, Ashley Jones, Justin Borevitz 2022. Plant assemble - Plant de novo genome assembly: annotation. protocols.io https://dx.doi.org/10.17504/protocols.io.kqdg36d87g25/v2Version created by Scott Ferguson
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it’s working
Created: March 20, 2022
Last Modified: March 21, 2022
Protocol Integer ID: 59655
Keywords: detailing plant genome assembly, plant genome assembly, protocols on multiple plant genome, correct plant genome, multiple plant genome, novo genome assembly, genome assembly, performing de novo assembly, plant assemble, sequencing technology, de novo assembly, genome, associated bioinformatics tool, novo assembler, gene homology, possible to de novo, rna, de novo, using oxford nanopore technology, oxford nanopore technology, protocol collection of bioinformatic workflow, complex plant
Abstract
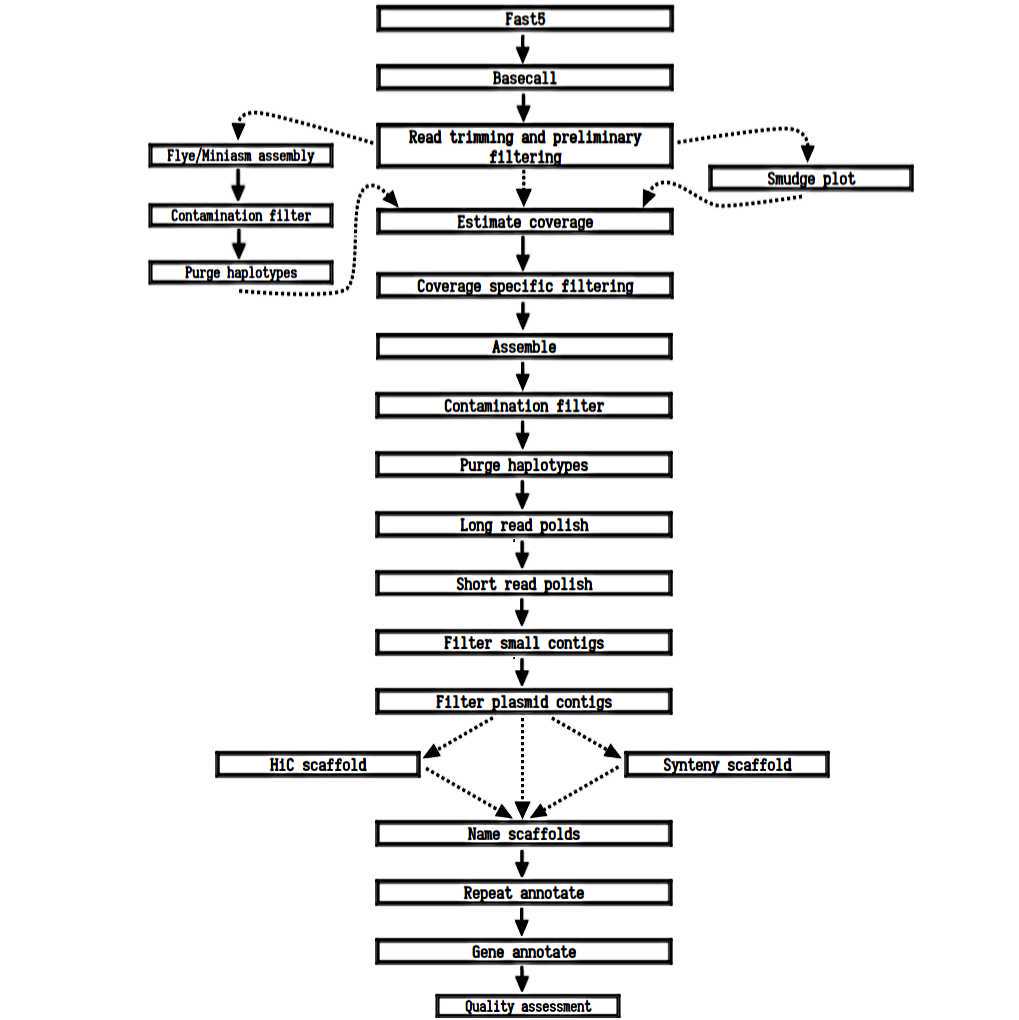
With the advancement of long-read sequencing technologies and associated bioinformatics tools, it has now become possible to de novo assemble complex plant genomes with unrivalled contiguity, completeness and correctness. As read lengths can surpass repeat lengths, the ability to assemble genomes de novo has dramatically improved, whereby complex plant genomes of widely variable sizes and repeat content have highly benefited. Despite these improvements, challenges remain in performing de novo assembly, namely in developing a reliable workflow and in tool choice. Here we present a protocol collection of bioinformatic workflows detailing plant genome assembly using Oxford Nanopore Technologies long-reads with a de novo assembler (Canu), syntenic or Hi-C scaffolding, and RNA and/or gene homology-based annotation. We have developed and tested these protocols on multiple plant genomes. Using these protocols with sufficient coverage of long-reads, a highly contiguous, complete, and correct plant genome can be assembled. These genomes can further genomic research into structural variation among groups, and SNP genotyping and association studies among populations.
Troubleshooting
Repeat annotate
Now that your genome is complete we want to find and annotate it for repeat regions. To do this we use EDTA and RepeatMasker. EDTA is first used to build a transposon (TE) sequence library. Next, RepeatMasker is used to find all TE and simple repeats within the genome.
Command
Repeat mask
Repeat annotate: soft mask
Output from ReapeatMasker will include a hard masked genome (all nucleotides within repeat regions will be changed to “N”) among other outputs. We will use the repeat regions defined within the out file and BEDTools to create a soft masked version of your genome (all nucleotides within repeat regions will be changed to lowercase, i.e. A becomes a, C becomes c, etc).
First, we create a bed file from the out file. Next BEDTools is used to soft mask our genome using the regions within the bed file.
Command
Soft mask
Gene annotate
Gene annotation can be done using RNA and/or gene homology. The tool we use is BRAKER2, which is capable of using both RNA and protein sequences of known genes. BRAKER2 will use your RNA or gene sequences to train a hidden markov model that scans your genome for regions likely to be genes. It will annotate both introns and exons, producing a gtf or gff3 file from which gene nucleotide and protein sequences can be made.
For gene annotation use a soft masked version of your genome, steps 1-2 (above).
Installing BRAKER2 can be hard and is very specific to your compute environment.
Gene annotate: Gene sequences/homology
From the NCBI (or another gene repository) find your species genus and download all gene amino acid sequences available. We also like to download the Arabidopsis thaliana genes and concatenate them to your genus's genes. Use this gene dataset for gene finding with BRAKER2.
Command
BRAKER2 homology
Gene annotate: RNA
Gene annotation with RNA begins by aligning your RNA to your genome. To do this a split read aware aligner is needed. The two current best aligners for this task are STAR and HISAT2 (https://daehwankimlab.github.io/hisat2/), both aligners give good results. We prefer to use STAR.
RNA alignment with STAR may benefit from dataset specific parameters. A very useful manual for STAR can be found on the STAR's github; https://github.com/alexdobin/STAR
Set “genomeSAindexNbases” to
Command
STAR - RNA alignment
Now that your RNA is aligned to your genome, we proceed to gene annotation with BRAKER2.
Command
BRAKER2 RNA
Gene annotate: Create protein sequences
Whether you did your annotation with RNA or gene homology, BRAKER2 will produce a gtf file which describes the location of genes within your genome (for a description of the gtf file format see: http://asia.ensembl.org/info/website/upload/gff3.html).
To create both amino acid and nucleotide sequences of your genes run the following:
Command
Create proetin sequences