Apr 15, 2023
Generating Sequencing Depth and Coverage Map for Organelle Genomes
- Yang Ni1,
- Jingling Li1,
- Chang Zhang1,
- Chang Liu1
- 1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College



Protocol Citation: Yang Ni, Jingling Li, Chang Zhang, Chang Liu 2023. Generating Sequencing Depth and Coverage Map for Organelle Genomes. protocols.io https://dx.doi.org/10.17504/protocols.io.4r3l27jkxg1y/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 15, 2023
Last Modified: April 15, 2023
Protocol Integer ID: 80567
Keywords: Reference genome, Paired-End Reads, Sequence Alignment, Sequencing Depth and Coverage, high assembly quality of organelle genome, high quality organelle genome, organelle genome, coverage map for organelle, assembly quality of organelle, sequencing depth, genome, organelle, sequencing, sequence depth, molecular marker, phylogenetic analysis, coverage map, coverage plot, assembly, assembly quality, errors in the assembly
Funders Acknowledgements:
CAMS Innovation Fund for Medical Sciences (CIFMS)
Grant ID: [2021-I2M-1-022]
National Science & Technology Fundamental Resources Investigation Program of China
Grant ID: [2018FY100705]
National Natural Science Foundation of China
Grant ID: [81872966]
Abstract
The success in development of molecular markers, phylogenetic analysis, and genetic engineering relies heavily on high quality organelle genomes. Any errors in the assembly of these genomes can result in inaccurate downstream analysis. In this article, we present a complete protocol for assessing the assembly quality of organelle genomes using sequencing depth and coverage plot. The protocol consists of nine steps that can be divided into three sections, allowing users to map sequence reads to the assembly, calculate sequence depth and coverage, and plot the data using a custom script, respectively. We provide detailed instructions for setting up the computational environment and running the analytical software tools. This protocol is particularly suitable for users with little bioinformatic experience and will play a vital role in ensuring the high assembly quality of organelle genomes.
Attachments

Guidelines
This protocol delineates the procedure for mapping raw paired-end reads to the reference genome utilizing the BWA aligner, processing the mapping data with SAMtools to compute the sequencing depth and coverage, and plotting the sequencing depth and coverage data using a custom python script. Examination of the sequencing depth and coverage map will allow investigators to evaluate the quality of organelle genome assembly and identify gaps or low-coverage regions. This knowledge aids in further assembly improvement and lays a solid foundation for downstream analyses.
Materials
All commonly used linux operating systems: CentOS and Ubuntu
Conda (version 4.7.12)
Sratoolkit (version 2.5.7)
BWA (version v0.7.17)
SAMTOOLS (version 1.16.1)
Troubleshooting
Before start
All commonly used linux operating systems: CentOS and Ubuntu
Conda (version 4.7.12)
Sratoolkit (version 2.5.7)
BWA (version v0.7.17)
SAMTOOLS (version 1.16.1)
Generating Sequencing Depth and Coverage Map for Organelle Genomes
1. Set up the working environment
1.1 Download the Miniconda installation script for Python 3.9 by running the following command in the terminal: (add the installation steps for the window platform!!!)
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
$ bash Miniconda3-latest-Linux-x86_64.sh -b -p ~/miniconda
$ export PATH="$HOME/miniconda/bin:$PATH"
$ conda create -n myenv python=3.9
$ conda activate myenv
#Please note that "$HOME" corresponds to the PATH you want to install in the conda environment. Here "myenv" is a custom name.
1.2 Install at least the following software:
$ conda install -c bioconda bwa
$ conda install -c bioconda samtools
# Installed the bwa and samtools.
$ wget -c https://anaconda.org/bioconda/sra-tools/2.11.0/download/linux-64/sra-tools-2.11.0-pl5321ha49a11a_3.tar.bz2# Downloaded the sra-tools 2.11.0 local package
$ conda install --offline -f sra-tools-2.11.0-pl5321ha49a11a_3.tar.bz2
# Installed the sra-tools 2.11.0 in offline mode. The offline installation was chosen because the conda networked installation of sratoolkits did not meet the decompression needs of this study.
1.3 Install the following software (Optional):
$ conda install -c bioconda -c conda-forge hisat2 star gmap tophat novoalign bowtie2
1.4 Install the python package if you need to visualize the sequencing depth and coverage map created with the script provided in this protocol (Optional).
$ conda install -c conda-forge numpy
$ conda install -c conda-forge pandas
$ conda install -c conda-forge matplotlib
2. Download the test data and reference genome
2.1 Download the reference genome (MT872375.fasta)
$ wget "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&id=MT872375&rettype=fasta&retmode=text" -O MT872375.fasta
# Download the reference genome with the accession number MT872375
2.2 Download the test data (SRR12597239)
$ wget -c https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-zq-38/SRR012/12597/SRR12597239/SRR12597239.lite.1
# Download the test data with the accession number SRR12597239
$ fastq-dump --split-files SRR12597239.lite.1
# Unzip the downloaded packet using sratoolkits
3. Create the index for the reference genome
# In the original directory
$ bwa index MT872375.fasta# Use BWA to create an index file for the reference genome
# Generate new .amb, .ann, .bwt, .pac, and .sa files
4. Align the raw paired-end reads to the reference genome and generate a SAM file
$ bwa mem -t 10 MT872375.fasta SRR12597239.lite.1_1.fastq SRR12597239.lite.1_2.fastq > out.sam
# Align using the BWA MEM algorithm, with -t 10 specifying 10 threads, input files are paired-end reads, output as out.sam
5. Convert the SAM file to a BAM file
$ samtools view -bS out.sam > out.bam
# Use samtools to convert the SAM file to a more efficient BAM format, -b for BAM output, -S for SAM input
6. Filter BAM data
$ samtools view -bF 12 out.bam > out.12F.bam
# Filter paired-end reads where both reads align to the reference genome (FLAG value of 12 indicates one of the paired-end reads did not align to the reference genome)
7. Sort the BAM File
$ samtools sort -@ 8 out.12F.bam -o sort_out.12F.bam
##Sort reads aligned to the reference genome, -o for specifying output file name, -T for temporary file name, -@ 0 for using 8 thread
#Sort paired-end reads where both reads align to the reference genome. The parameters are the same as those above.
8. Convert BAM data to TXT
$samtools depth sort_out.12F.bam > sort_out.12F.bam.txt
# Calculate the sequencing depth for paired-end reads where both reads align to the reference genome, output the results to a text file
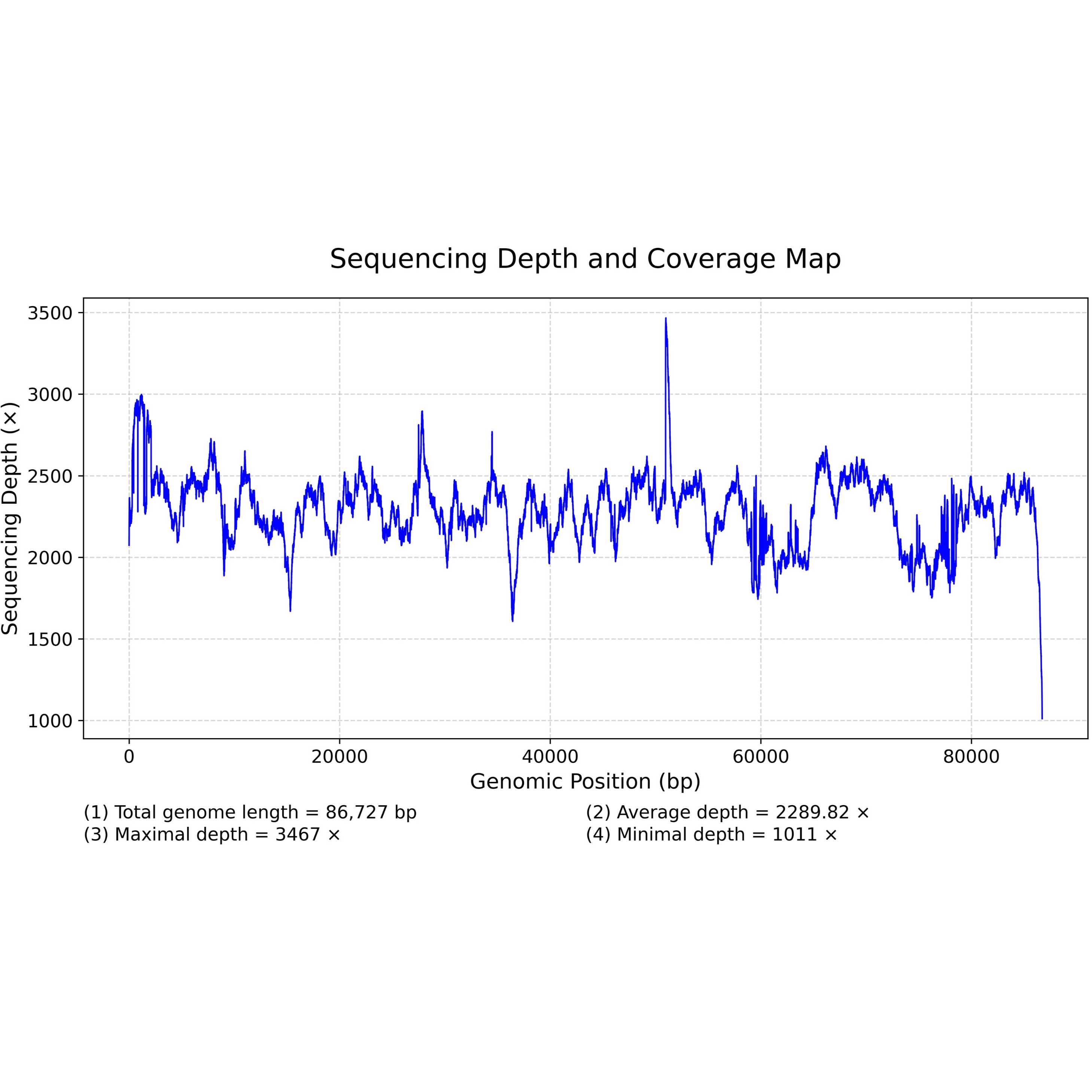
9. Draw the Genomic sequencing depth and coverage map
$ python Draw_SequencingDepth.py sort_out.12F.bam.txt Depth
# Input data: sort_out.12F.bam.txt; Output JGP format figure prefixation: Depth
# The Draw_SequencingDepth.py script was provided in the supplementary file 1. Before using this script, please install the python packages described in subsection 1.4. The final result is shown in Figure 1.