Apr 09, 2024
Biomarker's detection for diseases associated with metabolic disorder syndrome
- Cosme E. Santiesteban Toca1,
- Denisse Chacón2,
- Alejandro Rojo Moreno2,
- Saide Lizeth Medrano González3,
- Leyla Escalante Gonzalez4
- 1Tec Monterrey;
- 2Instituto Tecnológico y de Estudios Superiores de Monterrey;
- 3Tecnologico de Monterrey;
- 4Tec de monterrey
- Cosme E. Santiesteban Toca: Research and fulltime proffessor of engineering and science school campus Chihuahua;
- Denisse Chacón: Biotechnology Engineer;
- Alejandro Rojo Moreno: Biotechnology Engineer;



Protocol Citation: Cosme E. Santiesteban Toca, Denisse Chacón, Alejandro Rojo Moreno, Saide Lizeth Medrano González, Leyla Escalante Gonzalez 2024. Biomarker's detection for diseases associated with metabolic disorder syndrome. protocols.io https://dx.doi.org/10.17504/protocols.io.rm7vzxnb5gx1/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: June 19, 2023
Last Modified: April 09, 2024
Protocol Integer ID: 83650
Keywords: Machine learning, Biomarkers, Mellitus diabetes, Diagnosis and prognosis of diabetes, Genome assembly, Gene expression, Identification of genes, Functional annotation, Taxonomic annotation, Metabolic syndrome, Gut microbiota., gut microbiota, intestinal microbiota, identification of excretory protein, prediction of disease risk, biomarker potential for the diagnosis, biomarker potential, genes analysis, prognosis of diabetes, diabetes mellitus, excretory protein, microorganism, metabolic syndrome, gene expression, diabetes, bacteria, metabolic disorder syndrome, relationship with disease, disease, metabolic disorder syndrome the metabolic syndrome, associated disease, disease risk, data mining, machine learning, classification algorithm, prognosis
Funders Acknowledgements:
Tecnológico de Monterrey
Abstract
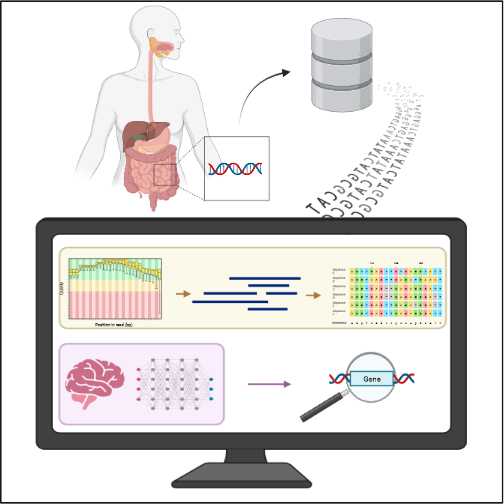
The metabolic syndrome (MetS) is known to substantially reduce the quality of life. MetS is associated with a high incidence of non-communicable diseases such as type 2 diabetes mellitus, cardiovascular diseases, cancer, among others. Multiple investigations focus the early diagnosis of MetS and its possible evolution in the patient on the basis of gene expression and clinical parameters.
However, we are interested in supporting the clinical diagnosis and prognosis of MetS-associated diseases based on the gut microbiota. Which means that we will take into account the set of microorganisms (bacteria, fungi, archaea, viruses and parasites) that reside in the intestine, given their relationship with diseases such as obesity, type 2 diabetes, as well as its influence on control glycemic.
Beyond of traditional diagnostic methods, Machine Learning (ML) can learn non-linear interactions iteratively from large amounts of data. This is possible using computer algorithms, which are already being applied in various fields, including the evaluation and prediction of disease risk.
The genes analysis belonging to the intestinal microbiota would allow the identification of excretory proteins with biomarker potential for the diagnosis and prognosis of diabetes and metabolic syndrome using supervised Machine Learning algorithms. For this reason, this project seeks to create a “pipeline” of classification algorithms (set of concatenated software) for data mining and analysis that allows predicting the appearance of type 2 diabetes and the progression of complications based on in the gut microbiota.
Guidelines
Main objectives:
- Analyze the intestinal microbiota of a patient.
~ Assembly of the intestinal genome.
~ Identification of the bacteria to which each genome belongs.
- Identify if the biomarkers that allow diagnosis and prognosis of diabetes and metabolic syndrome are present.
- Create a map that allows the identification of genes associated with microorganisms that may be involved in susceptibility or resistance to MetS-produced diseases.
- Learn automatically from the identified biomarkers to improve the diagnosis and prognosis of type 2 diabetes and obesity.
Experimental design:
Milestone 1: Access and download of the public databases in Amazon S3, of experiments of the intestinal microbiota associated with patients with type 2 diabetes. (1192 databases of between 4 and 12 Giga Bytes each).
- Download the SRA Toolkit
~ SRA official website: https://www.ncbi.nlm.nih.gov/sra
~ Anaconda: https://youtu.be/Q4jbA2UEP44
~ Github: https://github.com/ncbi/sra-tools
- Extract the FASTQ type files from the SRA access
~ “prefetch”: download the SRA files
~ “fasterq-dump”: extracts the FASTQ file
Milestone 2: Genome assembly from single-cell and multi-cell bacterial data. Being double read (forward & reverse), the assembly can generate up to four times the size of the original databases.
- Prepare the environment for experimentation.
- There are two methods for reading
~ Simple ending
~ Double ended
- Read the sequences.
- Extract the FASTQ file.
Milestone 3: Comparative analysis of the sequences obtained in milestone 2 with the sequences of each bacterium (BLAST).
- Download and configure BLAST+
- Download the BLAST BDs from:
- Perform the search with the filtered database
Milestone 4: Identify and segment genes with known functions and create a map that identifies them.
Troubleshooting
Before start
To facilitate bioinformatics processing in each step of the process, a group of specific tools are necessary:
Pipeline of bioinformatics tools
Download public databases
The SRA (Sequence Read Archive) is the standard format in which all NGS data is uploades into NCBI. To download and convert SRA files into FASTQ, download SRA Toolkit
Prepare the SRA-Toolkit workspace. For this step it is necessary to be located in the destination folder.
Command
Set the default folder to download sequences (Linux)
Download space
Command
new command name
Access and download public databases. In this case, a database from the human gut metagenome in Amazon S3 was used. "The gut microbiome related effect of Berberine and probiotics in treating Type 2 Diabetes" (NCBI Accession number PRJNA643353) is a database with 1192 datasets (4-12 GBs each) in consecutive order from SAMN15421765 to SAMN1522956 of experiments. The data were obtained from a randomized, double-blind, placebo-controlled trial on newly diagnosed type 2 diabetes patients from 20 centers in China where 409 patients were randomly assigned to receive BBR, probiotic with BBR, probiotics, or placebo for 3 months.
Use prefetch command followed by the number of the run from the desired experiment to download and create a folder with the archive in .sra format through SRA Toolkit. In this case, the BioSample SAMN15421765 is being used, which Run number is SRR12234739
Command
Downloads and creates a folder with the archive in .sra
prefetch
Extract FASTQ files from SRA access with fasterq-dump.
Command
Extract FASTQ files from SRA access
fasterq-dump
Quality control
NGS data can be affected by multiple reasons during the library preparations or the sequencing process, which can negatively impact the quality of the raw data. To perform quality control of the raw data download, download FASTX-Toolkit
Software
FASTX-Toolkit
NAME
Hannon Lab
DEVELOPER
REPOSITORY
Clean the sequences based on quality and size. Since there is no established consensus on the value these parameters should have, a value = >30 is assumed to determine good sequences.
Command
Removes sequences of low quality
fastq_quality_trimmer
Command
Removes sequences of low quality
fastq_quality_trimmer
Genome Assembly
Sequence reads from NGS consist of small genetic sequences much shorter than genomes and even genes. Thus, the assembly of these short sequences into larger sequences (contigs) is necessary. To perform the genome assembly of the reads, download Spades.
Software
Spades
NAME
Center for Algorithmic Biotechnology
DEVELOPER
REPOSITORY
SOURCE LINK
Read files with forward and reverse reads using -1 and -2 respectively
Command
Assembly
Assembly
Genome alignment
After the assembly, a reference genome is used to further piece together the sequenced data. Install BLAST setup for Unix to perform the Genome alignment
Download the reference databases (if necessary)
Command
Database download
Database download
Execute BLAST for nucleotides alignment
Command
blastn
blastn
Functional annotation
Now it is necessary to determine the biological function of the sequenced data. Install Prokka to perform functional annotation of the data.
Software
Prokka
NAME
Torsten Seemann
DEVELOPER
REPOSITORY
SOURCE LINK
Perform functional annotation
Command
prokka
prokka